4. ESMFold vs. AlphaFold2
In this module, you will use ESMFold to predict protein structures and systematically compare results with the AlphaFold2 predictions you generated in the previous module. ESMFold uses a fundamentally different approach—a protein language model trained on sequences alone, without requiring Multiple Sequence Alignments (MSAs) or templates. By comparing ESMFold to AF2, you will understand the trade-offs between different prediction methods and learn when to use each approach.
Live Workshop Session
Overview
ESMFold represents a different philosophy from AlphaFold2. While AF2 leverages evolutionary information through MSAs, ESMFold is built on a protein language model (ESM-2) that learns patterns directly from millions of protein sequences. This makes it:
- Much faster: No MSA generation step
- Better for orphan proteins: Works without homologs
- Simpler pipeline: Sequence in, structure out
But there are trade-offs—ESMFold may miss evolutionary insights that help AF2 achieve higher accuracy on well-studied proteins.
Learning Objectives
- Understand ESMFold’s language model approach vs AF2’s MSA-based approach
- Write Python code to use ESMFold following official documentation
- Compare prediction quality, speed, and confidence across different models
- Make informed decisions about model selection for different use cases
- Critically evaluate strengths and limitations of different prediction methods
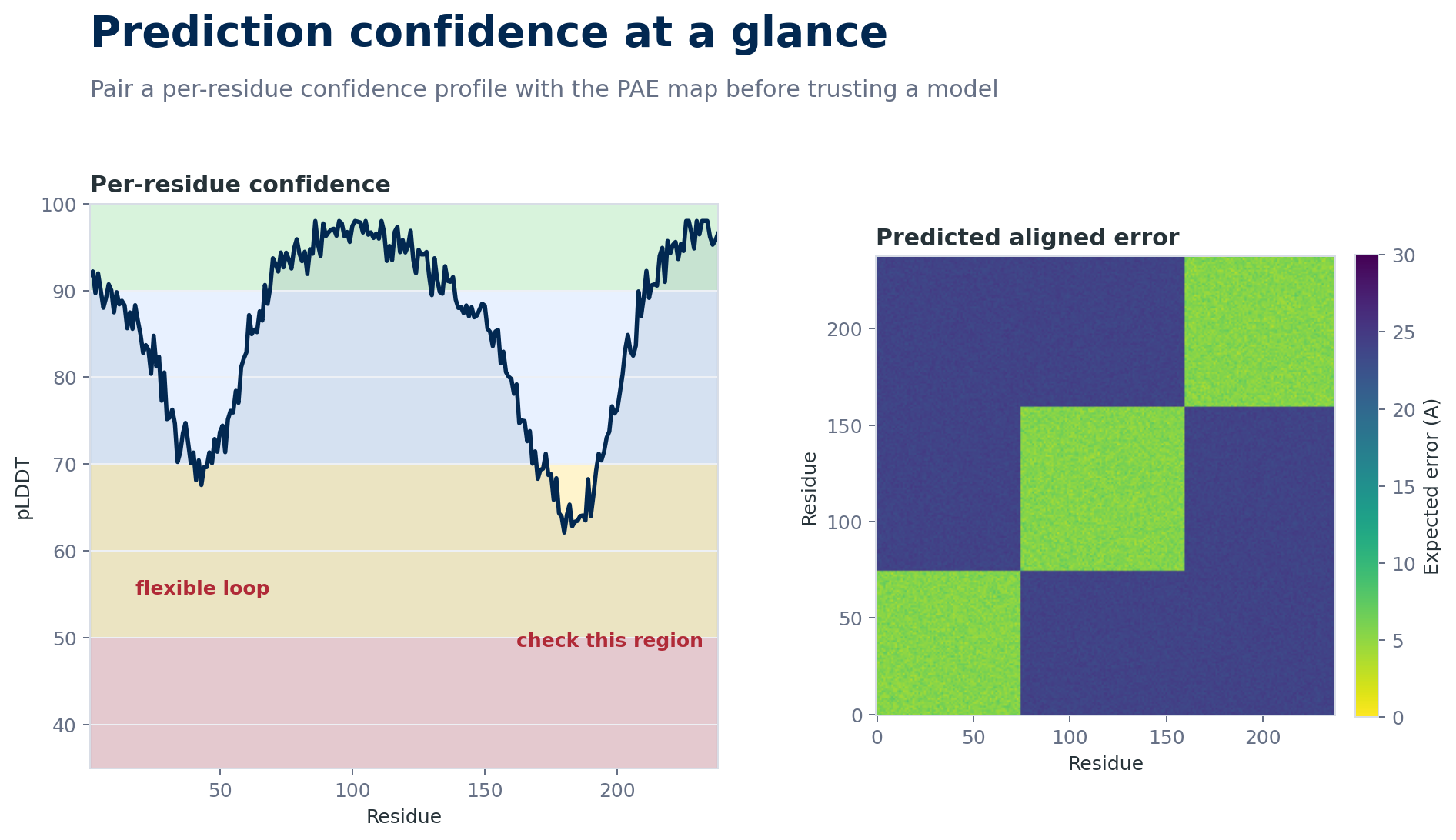
A useful structure-prediction review pairs residue-level confidence with pairwise uncertainty. High pLDDT alone is not enough if the PAE map says domains are uncertain relative to one another.
Hands-On Exercise
This module is structured as a hands-on comparison between ESMFold and AlphaFold2. You’ll run predictions, extract confidence scores, and develop intuition for when to use each tool.
Setup
Prerequisites: - Completed the AlphaFold2 module and have GFP predictions available - Access to a GPU (ESMFold requires ~8-16GB GPU RAM) - Python environment with PyTorch and the ESM library
Create your working directory:
mkdir esmfold_activity
cd esmfold_activityPart 1: Running ESMFold Structure Prediction
Goal: Generate an ESMFold prediction and understand the workflow.
1.1 Quick Introduction to ESMFold
Visit the ESM GitHub repository: - https://github.com/facebookresearch/esm
Key points about ESMFold: 1. Uses esm.pretrained.esmfold_v1() to load the model 2. Runs inference with model.infer_pdb(sequence) - returns a PDB-formatted string 3. Stores confidence scores (pLDDT) in the B-factor column of the PDB file 4. No MSA required - just sequence in, structure out!
1.2 Write Your ESMFold Prediction Script
Create a file predict_esmfold.py:
You can follow the official example from the ESM GitHub, or use this template:
import torch
import esm
import time
# Load model
print("Loading ESMFold model...")
model = esm.pretrained.esmfold_v1()
model = model.eval()
# Move to GPU if available
device = torch.device('cuda' if torch.cuda.is_available() else 'cpu')
model = model.to(device)
print(f"Using device: {device}")
# Your sequence (GFP - same as Activity 1)
sequence = "MSKGEELFTGVVPILVELDGDVNGHKFSVSGEGEGDATYGKLTLKFICTTGKLPVPWPTLVTTFSYGVQCFSRYPDHMKQHDFFKSAMPEGYVQERTIFFKDDGNYKTRAEVKFEGDTLVNRIELKGIDFKEDGNILGHKLEYNYNSHNVYIMADKQKNGIKVNFKIRHNIEDGSVQLADHYQQNTPIGDGPVLLPDNHYLSTQSALSKDPNEKRDHMVLLEFVTAAGITHGMDELYK"
# Run prediction with timing
print(f"Predicting structure for sequence of length {len(sequence)}...")
start_time = time.time()
with torch.no_grad():
output = model.infer_pdb(sequence)
elapsed = time.time() - start_time
print(f"Prediction completed in {elapsed:.1f} seconds ({elapsed/60:.2f} minutes)")
# Save to file
output_file = "esmfold_gfp.pdb"
with open(output_file, "w") as f:
f.write(output)
print(f"Structure saved to {output_file}")Questions to consider: 1. What does torch.no_grad() do? (Hint: disables gradient calculation - saves memory during inference) 2. Why don’t we need to generate MSAs like in AlphaFold2? 3. How long did your prediction take?
1.3 Run Your First Prediction
python predict_esmfold.pyExpected output: - A PDB file named esmfold_gfp.pdb should be created - Note the prediction time - this is much faster than ColabFold!
Part 2: Extracting and Comparing Confidence Scores
Goal: Extract pLDDT scores from both ESMFold and AlphaFold2 predictions for comparison.
2.1 Understanding pLDDT Storage in PDB Files
Both ESMFold and AlphaFold2 store pLDDT confidence scores in the B-factor column of PDB files.
PDB format (ATOM lines): - Columns 1-6: “ATOM” - Columns 7-11: Atom serial number - Columns 12-16: Atom name - Columns 17-20: Residue name - Columns 23-26: Residue number - Columns 31-54: X, Y, Z coordinates - Columns 61-66: B-factor (where pLDDT is stored!)
2.2 Extract pLDDT Scores
We’ve provided a script extract_plddt.py that works for both ESMFold and AlphaFold2 PDB files.
Run it on your ESMFold prediction:
python extract_plddt.py esmfold_gfp.pdbRun it on your AlphaFold2 prediction from the previous module:
python extract_plddt.py path/to/your/af2_prediction.pdbQuestions: 1. What is the average pLDDT for ESMFold vs AlphaFold2? 2. Which model has more high-confidence residues (>90)? 3. Do both models identify the same low-confidence regions?
2.3 Visualize Confidence Scores in PyMOL
Load your ESMFold structure and color by confidence:
load esmfold_gfp.pdb
# Color by B-factor (pLDDT): blue = high confidence, red = low confidence
spectrum b, blue_white_red, minimum=50, maximum=100Identify and display low confidence regions:
# Select residues with low confidence
select low_conf, b < 70
show sticks, low_conf
color yellow, low_confCompare to AlphaFold2 visually: - Do the same for your AF2 prediction - Are low-confidence regions in similar locations?
Part 3: Direct Comparison - ESMFold vs AlphaFold2
Goal: Compare the two predictions structurally and assess accuracy.
3.1 Prepare for Comparison
You need: 1. Your ESMFold prediction: esmfold_gfp.pdb (from Part 1) 2. Your AlphaFold2 prediction from the previous module (ColabFold output) 3. The experimental crystal structure of GFP from PDB
Download the crystal structure:
wget https://files.rcsb.org/download/1GFL.pdb -O 1gfl_native.pdb3.2 Visual Comparison in PyMOL
Load and align all three structures:
# Load all structures
load 1gfl_native.pdb, native
load path/to/af2_prediction.pdb, af2
load esmfold_gfp.pdb, esmfold
# Align both predictions to the native structure
align af2, native
align esmfold, native
# Display with different colors
hide everything
show cartoon
color green, native
color cyan, af2
color magenta, esmfoldAnalysis questions: 1. Do ESMFold and AF2 agree on the overall fold? 2. Where are the major differences (if any)? 3. Which one looks closer to the native structure? 4. Are differences in well-structured regions (beta-barrel) or loops?
3.3 Calculate RMSD
In PyMOL, calculate RMSD for each comparison:
Use the same approach you used in the AlphaFold2 module. If you’re feeling confident, try writing a PyMOL script to automate this!
# In PyMOL:
rms_print af2, native
rms_print esmfold, native
rms_print af2, esmfoldRecord your results:
| Comparison | RMSD (Å) | Interpretation |
|---|---|---|
| AF2 → Native | Better accuracy = lower RMSD | |
| ESMFold → Native | ||
| AF2 ↔︎ ESMFold | Agreement < 2Å is good |
Discussion points: - Which model is more accurate for GFP? - Do the two models agree with each other (RMSD < 2Å)? - Given that both are trained on different data, what does their agreement (or disagreement) tell you?
Part 4: Exploring ESMFold Features ⭐ OPTIONAL - If You’re Ahead
⏰ Due to time constraints, this section is optional. Only do this if you finish the core activity early, or come back to it later if you want to explore ESMFold more deeply!
Goal: Understand ESMFold-specific capabilities and parameters.
4.1 Memory Optimization with Chunking
ESMFold supports chunking to reduce memory usage.
Looking at the documentation, you’ll find model.set_chunk_size(size): - Reduces memory from O(L²) to O(L) - Useful for very long sequences - May be slightly slower
Modify your prediction script to test chunking:
# Before inference:
model.set_chunk_size(128)Experiment: 1. Run with no chunking 2. Run with chunk_size=128 3. Run with chunk_size=64
Questions: 1. Does chunking affect the output structure? 2. Does it affect runtime? 3. When would you use this?
4.2 Understanding Model Determinism
Test if ESMFold is deterministic:
Run the same prediction twice:
python predict_esmfold.py # Run 1
mv output.pdb output1.pdb
python predict_esmfold.py # Run 2
mv output.pdb output2.pdbCompare in PyMOL:
Expected result: RMSD = 0.0 Å (identical)
This tells us: - ESMFold is deterministic (same input → same output) - Unlike some methods with dropout, it’s reproducible - You can’t generate diversity by running multiple times
Compare to AF2: - AF2 gives 5 different predictions (different model weights) - Each can be slightly different - Provides ensemble diversity
Discussion: - Advantages of determinism? - Disadvantages (no ensemble diversity)? - How do you assess uncertainty with a single prediction?
Part 5: Speed Comparison - ESMFold vs AlphaFold2
Goal: Understand the practical speed differences between the two approaches.
5.1 Compare Prediction Times
You already timed your ESMFold prediction in Part 1. Now compare it to your ColabFold run.
Think back to your AlphaFold2 predictions: - How long did the MSA search take? - How long did the actual structure prediction take (for 5 models)? - What was the total time?
Fill in your observations:
| Model | Approximate Time | Notes |
|---|---|---|
| ESMFold | _____ seconds/minutes | From Part 1 output |
| ColabFold MSA search | _____ minutes | From AlphaFold2 module |
| ColabFold prediction (5 models) | _____ minutes | From AlphaFold2 module |
| Total ColabFold | _____ minutes |
Key observations: 1. ESMFold is typically 10-60x faster than full AlphaFold2 pipelines 2. Most of AF2’s time is spent on MSA generation (searching sequence databases) 3. ESMFold skips this entirely - it’s trained to predict from sequence alone
5.2 When Does Speed Matter?
Consider these scenarios and think about which model you’d choose:
Scenario 1: Screening 1,000 designed proteins for a protein engineering project - ESMFold could finish in hours - AF2 could take days or weeks - Which would you use first? Why?
Scenario 2: Final structure for an important publication - You need the highest accuracy possible - You have time for validation - Which would you use? Would you run both?
Scenario 3: Analyzing 50 point mutations of a therapeutic antibody - Need quick results to guide next experiments - Medium accuracy is acceptable for initial screening - Your strategy?
Remember: In practice, many researchers use ESMFold for fast screening, then validate interesting hits with AlphaFold2 for higher accuracy.
Part 6: Choosing the Right Model for Your Research
Goal: Develop intuition for when to use ESMFold vs AlphaFold2.
6.1 Key Differences Summary
| Aspect | ESMFold | AlphaFold2 |
|---|---|---|
| Training approach | Language model on sequences | Structure prediction with MSA+templates |
| Input required | Sequence only | Sequence (MSA search automatic) |
| Speed | Very fast (seconds-minutes) | Slower (minutes-hours with MSA) |
| Number of predictions | 1 (deterministic) | 5 (different model weights) |
| Best for | Fast screening, novel proteins | High-stakes accuracy, well-studied proteins |
| Needs homologs? | No | Performs better with many homologs |
6.2 Decision Guide
Use this guide to help choose the right tool:
START: I need to predict a protein structure
↓
Q: Is speed critical? (screening many sequences, quick answer needed)
YES → Use ESMFold first
NO ↓
Q: Is this a designed/synthetic protein with no natural homologs?
YES → Use ESMFold (AF2 needs homologs to work well)
NO ↓
Q: Is this high-stakes? (publication, drug design, experimental planning)
YES → Run BOTH models and compare
- Builds confidence if they agree
- Reveals uncertainty if they disagree
NO ↓
Q: Do I need uncertainty estimates from multiple predictions?
YES → Use AlphaFold2 (5 models provide ensemble diversity)
NO → Use ESMFold (faster, single high-quality prediction)6.3 Practical Scenarios - Test Your Understanding
For each scenario below, think about which model(s) you would use and why:
Scenario 1: Screening 5,000 designed protein variants for stability - Consider: Time constraints, cost, accuracy needs - Your choice: _______ - Why? _______
Scenario 2: Structure for molecular replacement in X-ray crystallography - Consider: Accuracy is critical, you need the best possible model - Your choice: _______ - Why? _______
Scenario 3: Novel metagenomic protein (no known homologs) - Consider: No evolutionary information available - Your choice: _______ - Why? _______
Scenario 4: Well-studied enzyme with 100+ homologs in PDB - Consider: Rich evolutionary information available - Your choice: _______ - Why? _______
Scenario 5: 200 point mutations to assess effect on protein binding - Consider: Need balance between throughput and accuracy - Your choice: _______ - Why? _______
6.4 What If the Models Disagree?
If you run both ESMFold and AlphaFold2 and they give different structures:
Step 1: Check confidence scores - Which model has higher average pLDDT? - Where specifically do they disagree (structured regions or flexible loops)?
Step 2: Understand the disagreement - Small differences (<2Å RMSD): Likely both correct, minor variations - Large differences (>4Å RMSD): Real uncertainty - investigate further
Step 3: What to do about it - If ESMFold has higher confidence → May indicate AF2 lacks good templates - If AF2 has higher confidence → Evolutionary information may be providing key insights - If both have low confidence in same region → Likely disordered or genuinely uncertain - When in doubt: Use predictions to design experiments, not as final answers
Remember: Predictions are computational hypotheses. They guide experiments but don’t replace them!
Part 7: Key Takeaways & Best Practices
7.1 What You’ve Learned
ESMFold: ✅ Very fast (no MSA generation needed) ✅ Works on designed/orphan proteins with no homologs ✅ Simple, streamlined workflow ✅ Great for high-throughput screening
⚠️ Single prediction (no ensemble diversity) ⚠️ May miss evolutionary insights ⚠️ Less extensively validated than AF2
AlphaFold2: ✅ Leverages evolutionary information (MSAs) ✅ 5-model ensemble for uncertainty estimation ✅ Extensively validated, highly accurate ✅ Best performance on proteins with many homologs
⚠️ Slower (MSA search takes time) ⚠️ Needs homologs for best performance ⚠️ Higher computational cost
7.2 Best Practices for Structure Prediction
Always do this: 1. ✅ Check pLDDT confidence scores - don’t trust low-confidence regions blindly 2. ✅ Inspect low-confidence regions carefully (may be disordered or uncertain) 3. ✅ Consider biological context (is this region expected to be structured?) 4. ✅ Validate critical predictions experimentally when possible
Smart strategies: 1. 💡 Use ESMFold for rapid screening → follow up interesting hits with AF2 2. 💡 For high-stakes work, run both models and compare 3. 💡 When models disagree, use that as a signal to investigate further 4. 💡 Low pLDDT doesn’t always mean “wrong” - might indicate genuine flexibility
7.3 Recommended Workflow for Most Projects
New protein structure question
↓
Start with ESMFold (fast initial prediction)
↓
Check pLDDT scores
↓
High confidence (>70) overall? ──NO→ Run AlphaFold2
↓ (may need evolutionary info)
YES Compare results
↓
Is this critical? ──YES→ Validate with AlphaFold2
↓ (get ensemble diversity)
NO
↓
Use ESMFold prediction
(fast, good enough for most purposes)The bottom line: There’s no single “best” model. Choose based on your specific needs for speed, accuracy, and the biological context of your protein!
Additional Resources
Documentation
- ESM GitHub: https://github.com/facebookresearch/esm
- ESMFold paper: Lin et al. (2023) Science 379:1123-1130
Online Resources
- ESMFold web server: https://esmatlas.com/resources?action=fold
- AlphaFold Database: https://alphafold.ebi.ac.uk/
- RCSB PDB: https://www.rcsb.org/